The integration of AI in drug discovery is revolutionizing the way researchers approach the development of new treatments for various diseases. Traditional methods are often time-consuming and costly, with the process of bringing a new drug to market taking up to 15 years and costing between $1–2B.
By using AI and advanced computational tools, researchers can now accelerate the identification of new drugs, significantly reducing both the time and cost involved in the drug discovery process.
Challenges of traditional drug discovery
In conventional drug discovery workflows, researchers first identify a biological target, such as a protein involved in disease progression, and then search for molecules that can modulate this target. The complexity of biological systems, combined with the vast number of potential chemical structures, estimated at around 1060, makes this a daunting task.
Traditional computer-aided drug discovery (CADD) methods often rely on simplified models and assumptions that fail to capture the intricacies of drug-target interactions, leading to high attrition rates in clinical trials.
An AI-driven approach to virtual screening
Innoplexus is a registered NVIDIA Inception startup. Their proprietary deep learning method uses NVIDIA NIM microservices to streamline the drug discovery process. They also use NVIDIA H100 GPU clusters featuring the following components:
- Accelerator: NVIDIA H100 Tensor Core GPU
- Memory: 80-GB HBM3 (High-Bandwidth Memory)
- Interconnect: NVIDIA NVLink 4.0
- Cluster configuration: Scalable, multi-node clusters with high-speed interconnects for distributed training and inference
This approach is informed by the NVIDIA NIM Agent Blueprint for generative virtual screening, which enables the rapid, AI-driven generation of novel molecular structures for accelerated molecular simulations and docking with NIM microservices.
Combining Innoplexus’ expertise with NVIDIA’s cutting-edge AI technology fundamentally transforms how innovative treatments are discovered and brought to market—making this process faster, more efficiently, and more precise.
To address the urgent need for novel therapies for neurodegenerative diseases associated with TDP-43 aggregation, Innoplexus developed an AI-driven drug discovery pipeline.
Innoplexus’s deep learning method
Innoplexus’ method employs custom-designed artificial neural networks (ANNs) for protein target prediction, trained on large-scale datasets of protein sequences, structural information, and molecular interactions.
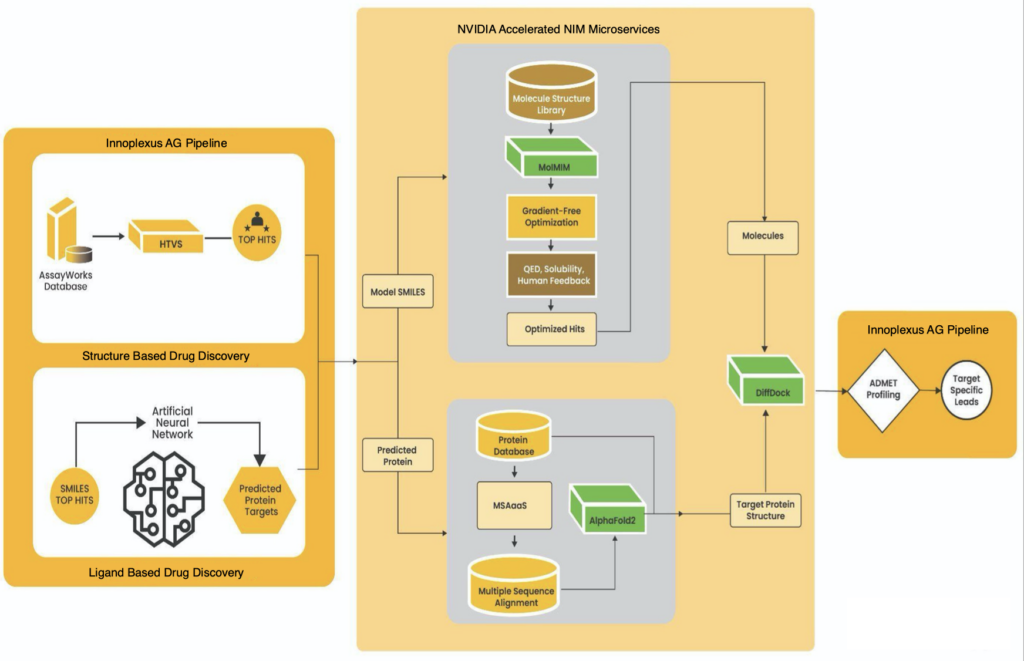
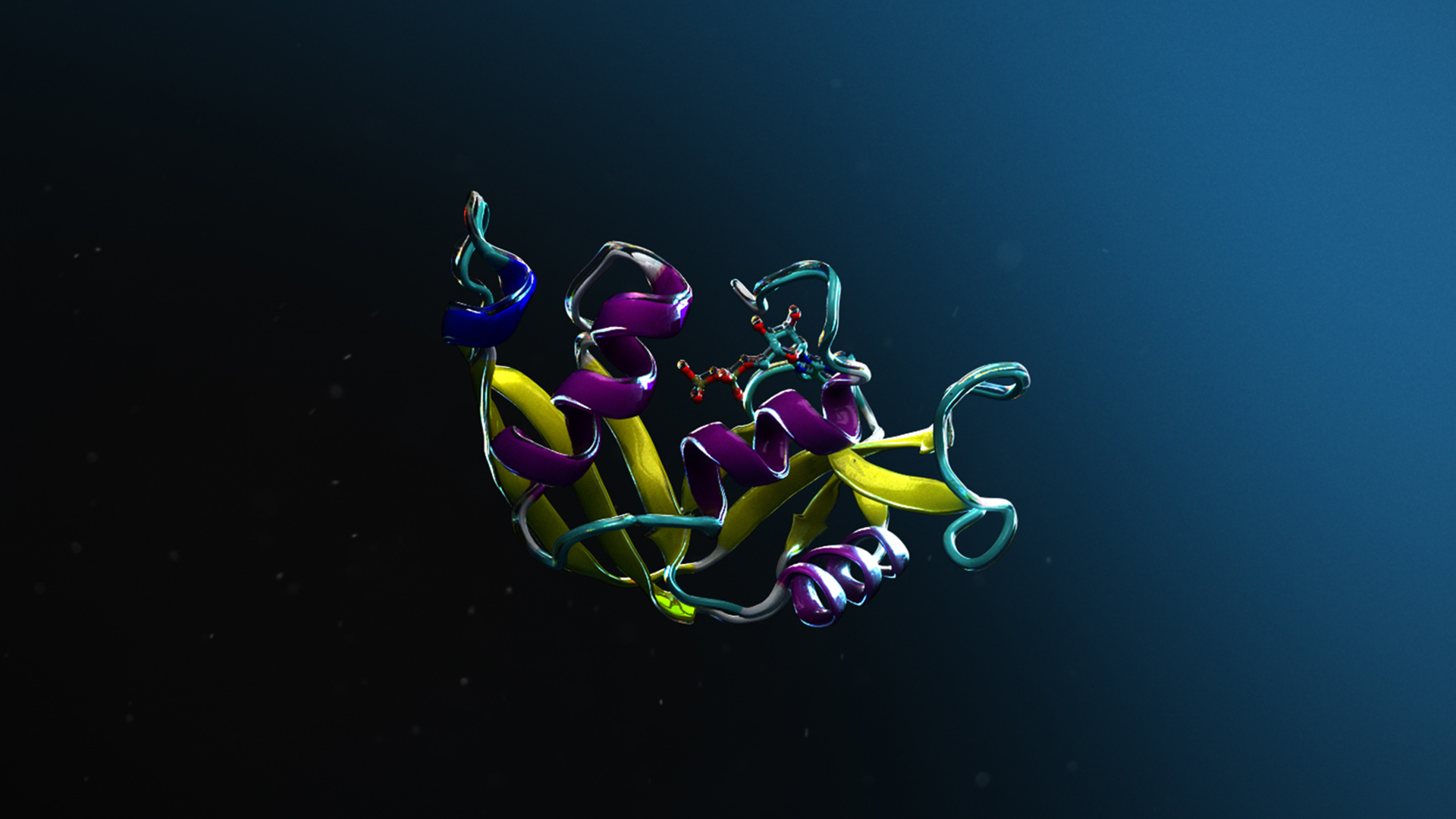
Figure 1. The workflow for structure and ligand-based drug discovery using NVIDIA NIM microservices
Innoplexus uses the following NVIDIA NIM microservices:
- AlphaFold2 for protein structure prediction
- MolMIM for optimized lead generation
- DiffDock for molecular docking
By combining these advanced AI tools, Innoplexus aims to streamline the drug discovery process and identify promising candidates that can effectively target TDP-43 and mitigate the progression of these debilitating diseases. This innovative approach has the potential to accelerate the development of new treatments and improve the lives of patients affected by neurodegenerative conditions.
AlphaFold2 for protein structure prediction
A protein sequence provided by the user is processed through the AlphaFold2 NIM microservice, which accurately determines the 3D structure of the target protein. This step involves aligning the sequence with known proteins, offering multiple alignment configurations for improved accuracy.
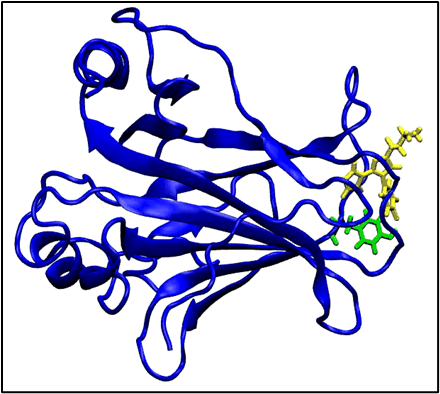
MolMIM for optimized lead generation
An initial chemical structure is passed through the MolMIM NIM microservice, which generates new molecular structures optimized for specific properties such as drug-likeness (QED), solubility (penalized log P), and molecular similarity.
The generated molecules are iteratively optimized in multiple cycles, depending on your requirements.
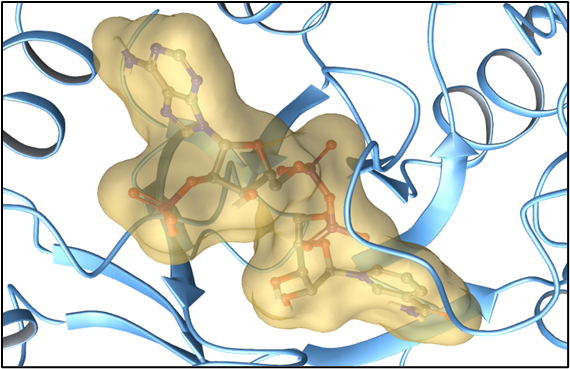
DiffDock for molecular docking
Molecular docking helps in determining the optimum site on the target protein where the drug binds. The optimized molecules and the target protein structure are processed by DiffDock, which predicts the binding poses of the molecules to the protein.
You can define the number of poses and other docking constraints, enabling a comprehensive analysis of potential drug-target interactions.
Post-processing ADMET pipeline
After DiffDock, the top 1K small molecules are further screened using the proprietary ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) pipeline, which assesses the pharmacokinetic and pharmacodynamic properties of the molecules.
This pipeline includes the following components:
- ADMET Prediction: The proprietary model predicts the ADMET properties of the molecules, including solubility, permeability, metabolism, and toxicity.
- Filtering and Ranking: Molecules are filtered and ranked based on their predicted ADMET properties, ensuring that only the most promising candidates are selected for further development.
Innoplexus ADMET model
The ADMET model is a custom-designed neural network that uses a large dataset of molecular structures and their corresponding ADMET properties.
The model is trained using advanced techniques:
- Multi-task learning: The model is trained on multiple ADMET tasks simultaneously, improving its overall performance and accuracy.
- Transfer learning: The model is fine-tuned on a large dataset of molecular structures, enabling it to generalize well to new, unseen molecules.
Workflow optimization
The pipeline is optimized for performance:
- Data parallelism: Distributed training and inference across multiple GPUs and nodes.
- Model parallelism: Splitting large models across multiple GPUs and nodes.
- Pipeline parallelism: Overlapping computation and communication between pipeline stages.
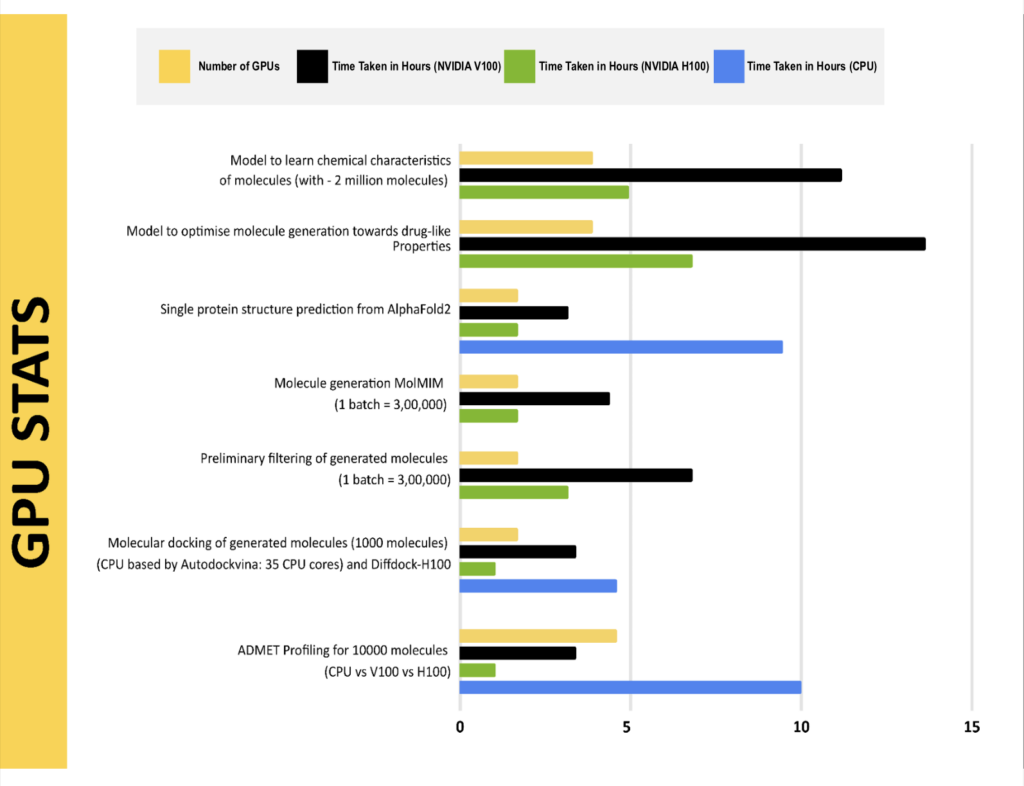
The application of GPUs and the approaches for accelerated computing facilitated in performing the compute-intensive operations of this solution fast, and making it feasible to complete within practical timelines.
Real-world applications and implications
Rapid compound identification with Innoplexus’AI-driven pipeline powered with NVIDIA H100 clusters, accelerates virtual screening of generated molecules in addition to molecular docking up to 10x, enabling researchers to perform the following tasks:
- Screen 5.8M small molecules in 5–8 hours duration.
- Identify the top 1% of compounds with high therapeutic potential from ADMET profiling in a few hours for a million compounds.
- Optimize lead compounds with 90% accuracy.
By harnessing the power of AI and high-performance computing, you can rapidly explore vast chemical spaces and pinpoint promising candidates for therapeutic development, significantly accelerating the drug discovery process.
Get started
AI and high-performance computing are set to transform the field of drug discovery, enabling faster, more accurate identification of potential drug candidates.
By combining cutting-edge neural network algorithms, generative models, and advanced molecular docking techniques, the Innoplexus virtual screening pipeline offers a powerful tool for accelerating the discovery of new drugs, ultimately improving patient outcomes and reducing the cost and time associated with bringing new therapies to market.
Get started with the NVIDIA NIM Agent Blueprint for generative virtual screening and learn more about Innoplexus.